Lumen is back
After many years the Lumen code is back.The new version of Lumen is based on Yambo 5.3.0. You can find our new code on GitLab at: https://gitlab.com/lumen-code/ Soon we will release a stable version of the code
After many years the Lumen code is back.The new version of Lumen is based on Yambo 5.3.0. You can find our new code on GitLab at: https://gitlab.com/lumen-code/ Soon we will release a stable version of the code

This year the Yambo Developers’ meeting will be online. In the morning of Friday 4, 11 and 18 December. It will be the occasion to discuss new developments and recent achievement with the Yambo code. Topics will range from: accelerating calculations in 2D systems, to Yambo-Aiida interface, validation of the code, robots, python postprocessing etc….. Read More
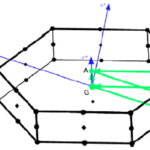
Here I show how to calculate GW correction for a generic k-point not included in the original grid used to perform a GW calculations using Yambo 4.2. This method allows to obtain the GW correction without the need of recalculate the full dielectric constant used in the initial calculation. As example I will take 2-dimensional… Read More
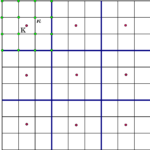
Convergence of dielectric constant with respect the number of k-points can be very slow in many systems. For this reason in Yambo it is implemented a double-grid technique that speeds up convergence versus k-points with a computational cost comparable to the standard calculations. The method is described in the paper: Speeding up the solution of… Read More
Here I present the BSE input file for yambo and I will discuss the meaning of each parameter. For a discussion on convergence issues see my post: Reasonable parameters for Yambo calculations. To generate this input type yambo -o b -k sex -y d -p p -V qp. I will not discuss the parameters relative… Read More
Example of a yambo input file for GW calculation in a solid. In order to generate the following input file you can use the command “yambo -g n -p p -V par” with yambo 4.0.2 (and with yambo 3.4.2 “yambo -g n -p p”). gw0 # [R GW] GoWo Quasiparticle energy levels ppa #… Read More

Here I report some advices to choose reasonable parameters in order to converge different kind of calculations with the Yambo code. The following parameters are a reasonable recommendation. However you have to remember that convergence can be different from system to system, so you should check your calculations and not use the following parameters blindly…. Read More
This is my small collection of Quantum Espresso (PWSCF) and Yambo input files that I used in my papers or for different testing purposes. I cannot give any warranty whatsoever these files are correct and fit your actual needs. Carbon nanotubes: 7×7, 4×2, 10×10, 1×10 Carbon nanoribbons: ANGR7 Diamond Donor Acceptor Complexes from ref. Appl. Phys. Lett…. Read More

My new tutorial on “Developing Yambo” is available online. This was possible thanks to the work of Myrta Gruning and Davide Sangalli that updated the tutorial to the last version of the code and completed some unfinished parts. In this tutorial we explain how to introduce new functionalities in the Yambo code. As example, inspired… Read More
Not working anymore in Yambo 4.5 but works in Yambo 5.x For molecules and also for molecular solids, the G0W0 approach often gives poor results. The main reason of this failure is the DFT starting point. In fact local or semi-local exchange correlation functionals give a too small gap compared with the experimental one, and… Read More