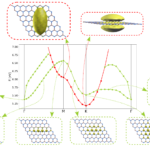
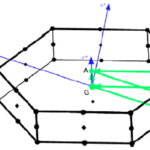
Yambo à la carte

A new talk on the Yambo code that I gave to the 1st TIMES School is available online: “Yambo à la carte“. In this talk I present the Yambo code and its functionalities. There is also a small presentation on the command lines, all the steps from DFT to Yambo and the installation.Hereafter the slides… Read More