Speed up dielectric constant calculations using the double-grid method with Yambo
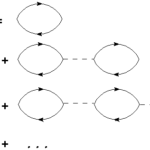
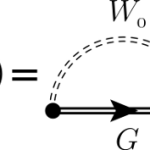
Convergence of dielectric constant with respect the number of k-points can be very slow in many systems. For this reason in Yambo it is implemented a double-grid technique that speeds up convergence versus k-points with a computational cost comparable to the standard calculations. The method is described in the paper: Speeding up the solution of… Read More