 When you work with experimentalists, often you have to deal with atomic structures reconstructed from x-ray scattering in the format of CIF files. Here I briefly explain how to transform these files in a QuantumEspresso input using Mercury code.
When you work with experimentalists, often you have to deal with atomic structures reconstructed from x-ray scattering in the format of CIF files. Here I briefly explain how to transform these files in a QuantumEspresso input using Mercury code.
Open the CIF file with Mercury then follow the points below:
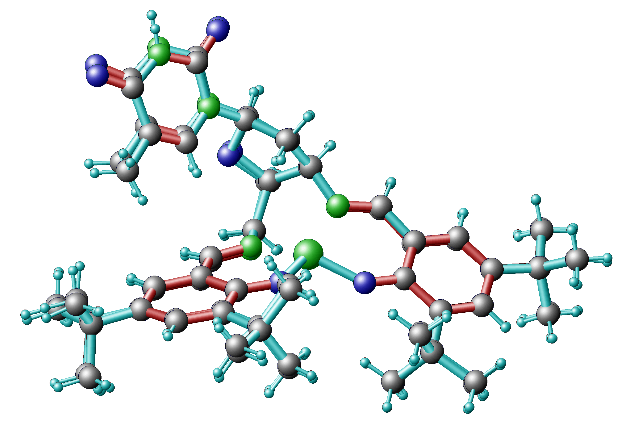
1) remove the duplicated atoms, the ones with fractional occupations, as for instance the blue atoms on top left of this figure. You just leave one atoms for each group and then you will optimize its position with DFT.
2) Go in the menu Calculate->Packing/Slicing active the flags Show cell axes and Pack between 0 and 1.0. On the right of the same windows you can decide how to pack atoms in the cell, I prefer the option “… in molecules whose Centroids fit” in such a way to maintain clear the molecular structure. Notice that sometime the packing gets confused if there are molecules at the border zone, in these cases you have to select a shorter packing range for example between 0.1 and 1.0.
3) Save atoms list from File -> Save As, notice that the units are Angstrom.
4) Get cell information from Display -> More Information -> Structure Information. Then translate it in the QuantumEspresso format. In order to do so you can put the namelist system the crystallographic constants a,b,c in Angstrom units and the angles as
cosAB = cos( γ ) [cosine of the angle between axis a and b]
cosAC = cos( β ) [cosine of the angle between axis a and c]
cosBC = cos( α ) [cosine of the angle between axis b and c]
and use ibrav=14.
5) Check your input file with Xcrysden with the command Xcryden –pwi input.in
and in particular that atoms distances and positions are the same of Mercury.
Other tools to translate CIF file to QuantumEspresso are available here: CIF to QuantumEspresso.