Theoretical Spectroscopy Lectures

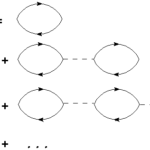
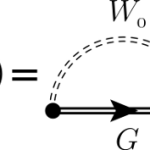
This year I was invited to give two lectures at the school on “Theoretical Spectroscopy” organized by F. Sottile, V. Olevano and G. Rignanese. I will present two lectures on “Non-linear approaches to TDDFT :: non perturbative real-time” and “Bethe-Salpeter Equation in real time” and a tutorial on the Lumen code. All the lectures are… Read More