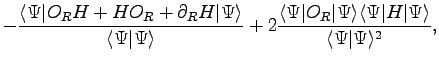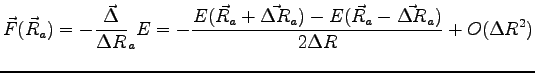In the last few years remarkable progresses have been made to develop
Quantum Monte Carlo (QMC) techniques which are able in principle
to perform structural optimization of molecules and complex systems
(52,29).
Within the Born-Oppheneimer approximation the nuclear positions
![]() can be considered as further
variational parameters included in the
set
can be considered as further
variational parameters included in the
set
![]() used for the SR minimization (2.13)
of the energy expectation value.
For clarity, in order to distinguish the
conventional variational parameters from the ionic positions,
in this section we indicate
with
used for the SR minimization (2.13)
of the energy expectation value.
For clarity, in order to distinguish the
conventional variational parameters from the ionic positions,
in this section we indicate
with ![]() the former ones, and with
the former ones, and with ![]() the latter ones. It is understood that
the latter ones. It is understood that
![]() ,
where a particular index
,
where a particular index ![]() of the whole set of parameters
of the whole set of parameters
![]() corresponds to a given spatial component (
corresponds to a given spatial component (![]() ) of the
) of the ![]() th ion.
th ion.
We computed
the forces ![]() acting on each of the
acting on each of the ![]() nuclear positions
nuclear positions
![]() , being
, being ![]() the total
number of nuclei in the system:
the total
number of nuclei in the system:
In order to evaluate the energy differences in Eq. 2.23 with a finite variance we have used the Space-Warp coordinate transformation (46,53). This transformation was also used in the evaluation of the wave-function derivatives respect to nuclear positions ![]() .
Even if Space-Warp transformation is a very efficient technique to reduce the variance of the forces, it is very time consuming and so for larger systems we preferred to use Zero Variance forces (29), as it was described in the chapter 1.
.
Even if Space-Warp transformation is a very efficient technique to reduce the variance of the forces, it is very time consuming and so for larger systems we preferred to use Zero Variance forces (29), as it was described in the chapter 1.
The ![]() operators are used
also in the definition of the reduced matrix
operators are used
also in the definition of the reduced matrix ![]() for those elements depending on the variation with respect to a nuclear
coordinate. In this way it is possible to optimize both the wave function
and the ionic positions at the same time,
in close analogy with the Car-Parrinello(54) method applied to
the minimization problem. Also Tanaka (48) tried to perform
Car-Parrinello like simulations via QMC, within the less efficient steepest
descent framework.
for those elements depending on the variation with respect to a nuclear
coordinate. In this way it is possible to optimize both the wave function
and the ionic positions at the same time,
in close analogy with the Car-Parrinello(54) method applied to
the minimization problem. Also Tanaka (48) tried to perform
Car-Parrinello like simulations via QMC, within the less efficient steepest
descent framework.
![\includegraphics[width=\textwidth]{Li2.eps}](img274.png) |
An important source of systematic errors is the dependence of the
variational parameters ![]() on the ionic configuration
on the ionic configuration ![]() ,
because for the final equilibrium geometry
all the forces
,
because for the final equilibrium geometry
all the forces ![]() corresponding to
corresponding to ![]() have to be zero, in order to guarantee that the true minimum
of the potential energy surface (PES) is reached (55,56).
As shown clearly in the previous subsection, within a QMC approach
it is possible to control this condition by increasing systematically
the bin length, when the thermal bias
have to be zero, in order to guarantee that the true minimum
of the potential energy surface (PES) is reached (55,56).
As shown clearly in the previous subsection, within a QMC approach
it is possible to control this condition by increasing systematically
the bin length, when the thermal bias ![]() vanishes.
In Fig. 2.3 we report the equilibrium distance
of the Li molecule as a function of the inverse bin length,
so that an accurate evaluation of such an important quantity
is possible even when the number of variational parameters is rather large,
by extrapolating the value to an infinite bin length.
However, as it is seen in the
picture, though the inclusion of the 3s orbital in the atomic AGP basis
substantially improves the equilibrium distance and the total energy by
vanishes.
In Fig. 2.3 we report the equilibrium distance
of the Li molecule as a function of the inverse bin length,
so that an accurate evaluation of such an important quantity
is possible even when the number of variational parameters is rather large,
by extrapolating the value to an infinite bin length.
However, as it is seen in the
picture, though the inclusion of the 3s orbital in the atomic AGP basis
substantially improves the equilibrium distance and the total energy by
![]() , this larger basis makes our simulation less efficient,
as the time step
, this larger basis makes our simulation less efficient,
as the time step ![]() has to be reduced by a factor three.
has to be reduced by a factor three.
We have not attempted to extend the geometry optimization to the more accurate DMC, since there are technical difficulties (57), and it is computationally much more demanding.