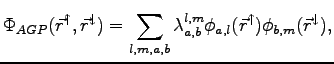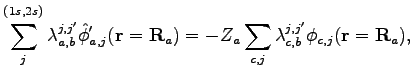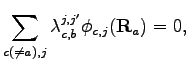Recently a new trial function was proposed for atoms, that includes both the terms (2). In the first part of this thesis we extend this promising approach to a number of small molecular systems with known experimental properties, that are commonly used for testing new numerical techniques.
The major advantage of this approach is the inclusion of many CI expansion terms with the computational cost of a single determinant. For instance this has allowed us to perform the full structural optimization of benzene without a particularly heavy computational effort on a single processor machine.
For an unpolarized system containing ![]() electrons
(the first
electrons
(the first ![]() coordinates are referred to the up spin electrons)
the AGP wave function is a
coordinates are referred to the up spin electrons)
the AGP wave function is a
![]() pairing matrix
determinant, which reads:
pairing matrix
determinant, which reads:
| (1.18) |
An important property of this formalism is the possibility to describe explicitly resonating bonds present in many structures, like benzene. A
![]() different from zero represents a chemical bond formed by
the linear combination of the m-th and n-th orbitals belonging
to a-th and b-th nuclei.
It turns out that resonating bonds can be well described through the geminal
expansion by switching on the appropriate
different from zero represents a chemical bond formed by
the linear combination of the m-th and n-th orbitals belonging
to a-th and b-th nuclei.
It turns out that resonating bonds can be well described through the geminal
expansion by switching on the appropriate
![]() coefficients: the
relative weight of each bond is related to the amplitude of the corresponding
coefficients: the
relative weight of each bond is related to the amplitude of the corresponding ![]() .
.
Also polarized systems can be treated within this framework, by using the spin generalized version of the AGP (GAGP), in which also the unpaired orbitals are expanded as well as the paired ones over the same atomic basis employed in the geminal (34).
Another important property of AGP wave-function is the size consistency:
if we smoothly increase the distance between two regions ![]() and
and ![]() ,
each containing a given number of atoms, the many-electron
wave function
,
each containing a given number of atoms, the many-electron
wave function ![]() factorizes into the product of space-disjoint
terms
factorizes into the product of space-disjoint
terms
![]() as long as the interaction between the electrons coupling the
different regions
as long as the interaction between the electrons coupling the
different regions ![]() and
and ![]() can be neglected. In this limit the total energy
of the wave function
approaches the sum of the energies corresponding to
the two space-disjoint regions. This property, that is obviously valid
for the exact many-electron ground state, is not always fulfilled by
a generic variational wave function as for instance configuration-interaction (CI) wave-function.
Notice that this property is valid when both the compound and the separated fragments have the minimum possible total spin. This is precisely the relevant case for hydrogen phase diagram studied in this thesis because we have not studied ferromagnetic or partially ferromagnetic phases, that are not believed to be present in the reasonable pressure-temperature range of hydrogen (for a discussion about ferromagnetism in high-pressure hydrogen see Ref. (35)).
can be neglected. In this limit the total energy
of the wave function
approaches the sum of the energies corresponding to
the two space-disjoint regions. This property, that is obviously valid
for the exact many-electron ground state, is not always fulfilled by
a generic variational wave function as for instance configuration-interaction (CI) wave-function.
Notice that this property is valid when both the compound and the separated fragments have the minimum possible total spin. This is precisely the relevant case for hydrogen phase diagram studied in this thesis because we have not studied ferromagnetic or partially ferromagnetic phases, that are not believed to be present in the reasonable pressure-temperature range of hydrogen (for a discussion about ferromagnetism in high-pressure hydrogen see Ref. (35)).
Now we want to highlight how it is possible to implement nuclear cusp condition (see Appendix C) for molecular systems with a pairing wave-function.
A straightforward calculation shows that the AGP wave function fulfills the
cusp conditions around the nucleus ![]() if the following linear system is
satisfied:
if the following linear system is
satisfied: